The End (Regretfully Without Commentary from Far Away Tropical Islands…)
Hello All,
I wished to be writing this from some far away tropical resort, as I was told PhD students of the past used to do before they defended their theses. Instead, this comes straight from the ROC homeland, deep in the midst of a global pandemic and a tea-fueled thesis writing session filled with Microsoft Word formatting errors and cat fur. As I see it, this is likely to be the last post that I contribute to the blog, as my defense is slightly over a month away and post that date, I will be moving to the land of ‘chow-dah’ and Dunkin. So this is it. In the words of the great John F. Kennedy: “This is the end. Hold your breath and count to ten. Feel the Earth move and then, hear my heart burst again. For this is the end.”
Y’all are gonna miss my colorful commentary, ain’t ya? Let’s get to it.
So, somewhere around February, we left off at the co-localization of 5637 bladder cancer extracellular vesicles (EVs) and a tetraspanin called CD81. Now this CD81 fella was a well-known general marker of EVs that was used extensively in the literature for demonstrating that the vesicles belonged to a secretive order called exosomes. Now this wasn’t their only mark of identification, no. Many were required to demonstrate proof that a vesicle belonged to this secretive order, but CD81 was a very common sign (well, published literature and our analysis told us that only ~ 15% of the order actually carry CD81 on them, but still let’s generalize a population, shall we?). So that was cool. We were able to show that our cancer-derived vesicles had this general marker. But since this was a general marker, what did that tell us about cancer? Well, to find out what these vesicles could indicate about cancer, we had to switch to a new identifier.
Now, the original marker that we were interested in for the barcoding of our misfit particles was a little protein called mucin 2 or MUC2. Now, a group of great scientists had previously demonstrated that EVs derived from bladder cancer cell lines (in particular the 5637 cell line) potentially expressed this protein at higher levels than healthy, bladder epithelium-derived EVs. This meant that it was possible that MUC2 would be a good indicator of the presence of bladder cancer in a urine sample. If an upregulation of MUC2 was found in the EV population, then that could mean that a patient was in for a very fun ride. So the journey began to hunt down these MUC2 expressing mother-flippers and get them to tell us whether or not they carried messages of bladder cancer. (Spoiler alert: our efforts will be spoiled.)
First, in order to do that, we had to prove that we could do a little something called particle counting. Now, I’ve also talked about this before (probably close to a year ago), but basically we use the wizardry of the great power ImageJ and his apprentice the plugin ComDet. This powerful duo allowed us to take confocal images (remember when I used to do SEM?) and count and co-localize the little dots that we saw. Now, during the analysis of the 5637 EVs and CD81, we observed that at high concentrations of vesicles caused a breakdown in the ability of the plugin to effectively count the particles on the membrane surface. And the major reason that this was occurring was because of a little thing called resolution. Now, the resolution of an imaging technique such as confocal microscopy is actually pretty high, much better than that of the standard epifluorescence imaging method. However, even it has its limitations and particles that are quite close together are next to impossible to resolve as two (or multiple) distinct particles. In fact, as we can see in Figure 1, which shows simulated particles at different imaging resolution levels (thanks to Baturay, who I hope is quite enjoying a well-deserved break with his fiancé in Germany).
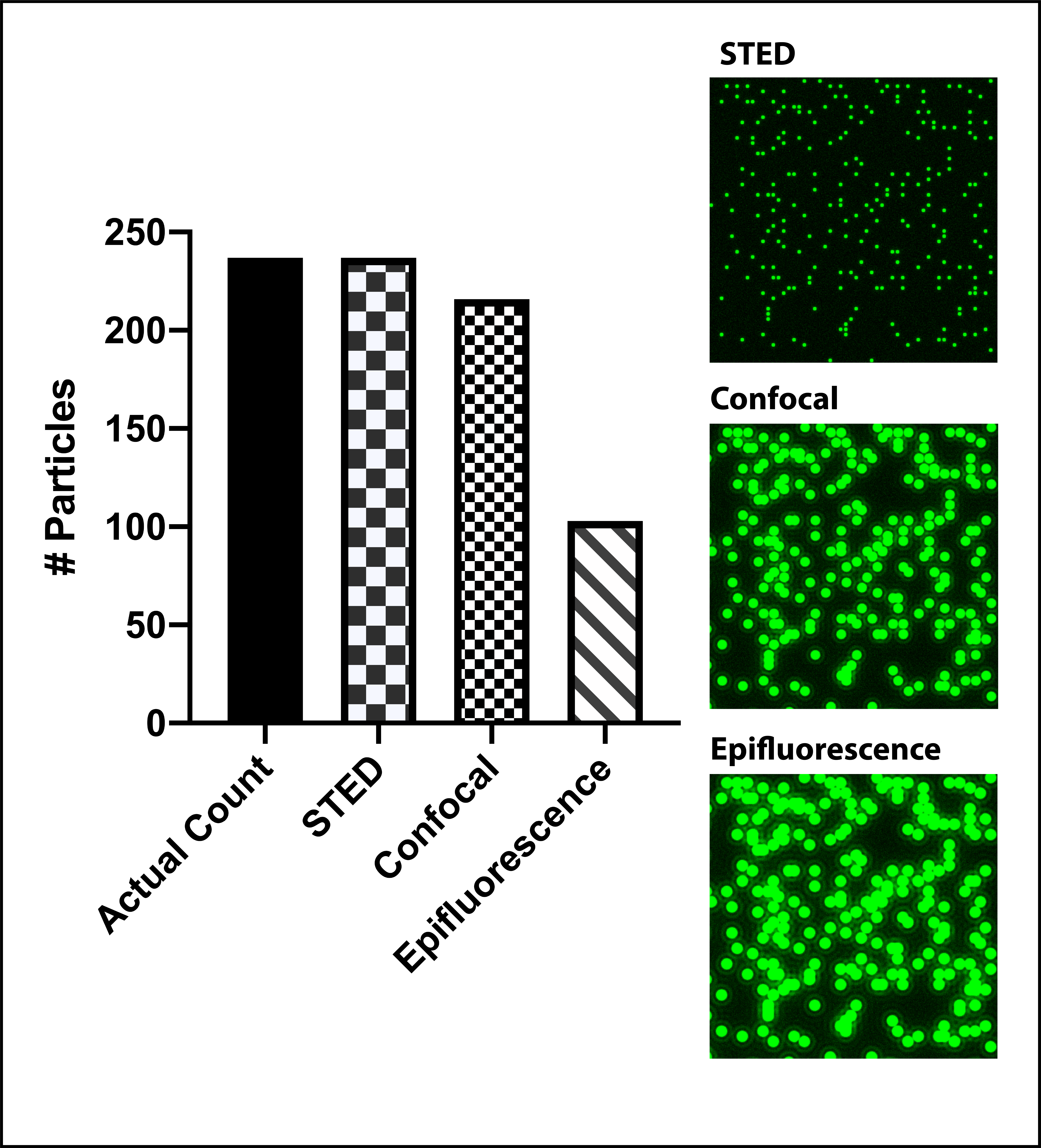
Figure 1: Simulated particle data showing the effect of imaging technique on counting particles.
Now in this figure, a field of ~250 particles is only accurately counted by a super resolution imaging technique. Confocal imaging provides a slightly worse count, as particles close together start to become indistinguishable from one another (I know, I know Bill, we do have deconvolution that could potentially, albeit computationally, solve this problem at the right concentrations) and epifluorescence imaging really soils the sheets in its ability to separate closely associated particles. While this data would suggest that the only real way to accurately count all the captured particles would be to use a super resolution microscope, this isn’t really practical as it actually takes quite a while to collect images by this method, the samples must be fixed in a mounting media, which leads to membrane bowing or breakage and it’s only really sensitive to certain wavelength dyes (looking at you here Red (Hellboy reference anybody)). That’s not to say that it can’t be used at all and we did in fact show that ability to distinguish close particles captured on a membrane (Figure 2).

Figure 2: TetraSpeck beads captured on the membrane. (A) Confocal and (B) STED.
Since we said that super resolution requires too much effort to make it a feasible imaging technique for a diagnostic, let’s talk about how we decided to approach counting particles on our fancy confocal system. Since the problem appears to be associated with high particle densities, we thought ‘well, what if we count at lower particle concentrations?’ Controlling the concentration of particles is something that is possible with known input concentrations (I mean not really practical for a straight up biological sample, but hey, the science has to start somewhere, right?). So we set out to test different dilutions of our fluorescent nanoparticles to see exactly how low we have to go to get decently accurate counting with out plugin. We varied the particle concentration from 1e8 particles/mL down to 1e5 particles/mL and captured them on the membrane. We then imaged the samples on confocal and extrapolated the total number of particles captured on the membrane and compared it to the expected capture. This was all performed in triplicate and it gave us the beautiful mess that is Figure 3.

Figure 3: Expected particle capture compared to experimental capture and analysis of TetraSpeck samples.
Okay, it’s not THAT much of a mess, but yeah it wasn’t great. Basically, this analysis showed us that for particle concentrations above 1e6 particles/mL, we couldn’t really accurately count the total captured particles. And you can see this by looking at the detection regions of interest (ROI) shown in the confocal images to the right of the graph. Basically, as the particles start piling on top of each other at high particle concentrations, it’s impossible to detect them, just like we predicted with the simulated data. Well, that sucks. Basically, in order to accurately interpret data, we have to operate at input concentrations of 1e6 particles/mL or less.
Well, that’s good to know. We just have to make sure that we are diluting our sample to be at this concentration or less. No biggie, easy peasy. That is, if you know the starting concentration of your vesicles. Luckily for us, we do know this value and it’s something that we could work with. Thus, with that knowledge in hand, we set out to try and count our vesicles and look for that dastardly MUC2. So we took some EVs from our bladder cancer cells and our healthy PBE cells and labeled them with both CFSE and an anti-MUC2 antibody. Excitedly, expecting a pretty similar result to the CD81 data, we ran the samples and went to image. Well, let’s just say that things didn’t exactly go as planned. Inconceivable! (Yes, I do know the meaning of that word and I do believe that I am using it correctly.)

Figure 4: MUC2 labeling of 5637 and PBE EVs. Despite the signal in the MUC2 channel, there was virtually no co-localization of the two labels, with the MUC2 antibody seemingly labeling background.
Cue me in the pose of Rodin’s The Thinker. Yeah. Not good. It looks like the protein that we were after didn’t seem to want to be found in these vesicles. Now, Jon did suggest that the specific epitope that the antibody targeted could be internalized, but a permeabilization test of the vesicles gave the exact same result: no dice. While this was disappointing news, we have a possible explanation as to why MUC2 might not be ideal for being a vesicle associated cancer marker. That idea is that it is possible that the protein could in fact be loosely associated with the surface of the vesicles and not actually found within the vesicular membrane. Because the initial data for MUC2 being a good marker for bladder cancer was based on a Western blot of an EV sample, it is highly possible that the protein co-precipitated with the vesicles during centrifugation and thus labeled the vesicles positive for MUC2. While this wasn’t ideal, it also wasn’t really the end of the world. There are other markers available, they just happen to be more general cancer markers.
So to salvage this listing ship, we decided to go after another marker that was known to be found in the vesicle membrane and also highly expressed in cancer: programmed death ligand 1 or PD-L1. Now, PD-L1 is well reported in the literature as a positive indicator of cancer because of its effects as a deterrent of the immune process of trying to detect and remove cancerous cells (tumor cells express it basically to hide themselves). So we thought ‘can we label the 5637 EVs for PD-L1 instead of MUC2 and observe a difference from the PBE vesicles? Well, all good stories don’t have sad endings, so this one must have a happy ending (I’ll let you have that one).
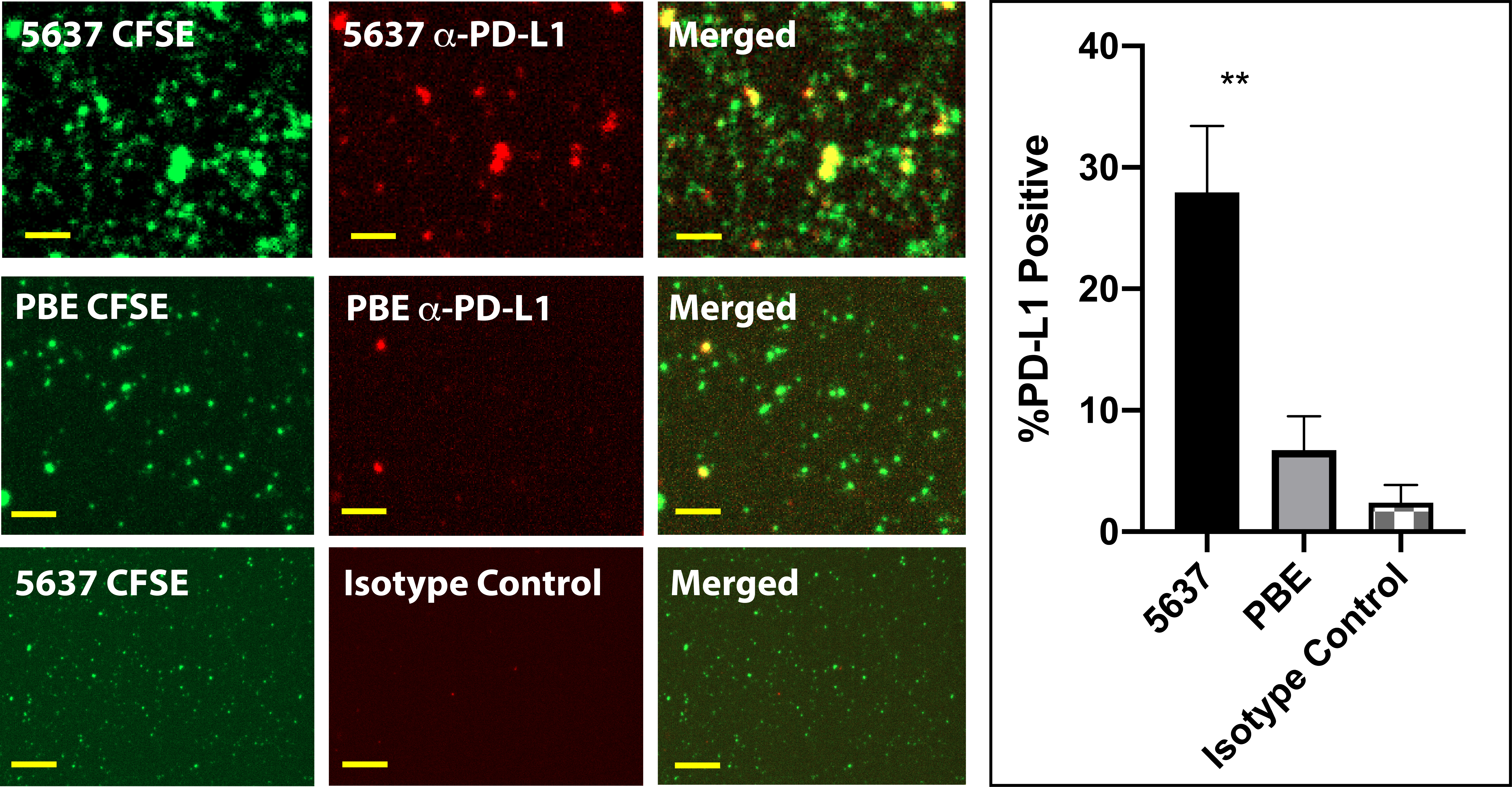
Figure 5: PD-L1 labeled EVs from 5637 cells, PBE and an isotype control. 5637 EVs express the PD-L1 protein at a significantly higher level than healthy cells. The isotype control indicates a minimal amount of non-specific labeling.
Yeah, that’s what I’m talking about! Nice, sexy data. Great statistics. Low non-specific labeling. All around not a bad show. Yeah, so it looks like PD-L1 is a good indicator of cancer on extracellular vesicles. I mean, this was already known, but hey, we were able to show that in our system. It’s actually kinda remarkable because you can sometimes even see vesicles that are right next to each other that do/don’t have the label. Pretty cool, right? I had even one more trick up my sleeve: Labeling for multiple targets on the same vesicle population. Remember that CD81 data from so many words ago? Yeah, I decided to try labeling for that at the same time as PD-L1. And damn were the results impressive.

Figure 6: Triple labeling of 5637 EVs with CFSE, anti-CD81 antibody and anti-PD-L1 antibody.
Yeah, that’s right there’s three distinct vesicles each with their own phenotype. One vesicle doesn’t express either CD81 or PD-L1, one vesicle expresses only PD-L1 and one vesicle expresses PD-L1 and CD81. Real neat, right?
Well, how do we best summarize this story and make it all into a format that makes sense? Well, let’s start with this: studying a population of vesicles looking for a “rare” vesicle that is one of a kind and is the only vesicle that expresses a particular biomarker may end up being a very, very difficult prospect. More than likely, it will take looking for an upregulation of a certain biomarker versus a control (healthy) vesicle population that tells us the result that we want. Kinda similar to the approach of using PSA for the detection of prostate cancer. There, you don’t really say “hey, you have this stuff, you have cancer” but rather “this stuff is starting to be expressed at higher than baseline levels and you might have cancer”. That may turn out to be the approach with trying to do a vesicle-based diagnosis of disease.
Furthermore, it is possible that a large number of the proteins that are suspected to be vesicle-associated and thus indicative of a disease when seen on a vesicle, may in fact be loosely associated proteins that are co-purified with the vesicles and will thus show up on a protein gel analysis of the vesicles. However, in this method of catch and display diagnostics, where the sample is captured on the surface and then analyzed, it is likely that this association disappears and we see that those proteins are not in fact found in the vesicle membrane and may not be good immunocytochemistry indicators of disease.
So that’s it. We have a platform that can allow us to study vesicles at the single vesicle resolution and determine whether or not certain proteins are good choices to target for immunocytochemistry approaches to disease diagnosis. That sums up everything that I have done in 5 years. We did it. I did it. It works, not perfectly and not exactly in the way that we intended, but it works.
Finally, in the words of the famous Nelson Mandela: “On that bombshell, good night!”