Adsorption Quantification VII
I realized last week that I had made a pretty substantial mistake with my quantification which involved my mass standards. Half way through running the gels for the previous study I had accidentally started making my mass standards at a higher concentration but counting them in the quantification as their initial value. The bottom line is that I was underestimating the amount of adsorption on the samples, which makes sense given the anomalous crystalline silicon adsorption the I received most recently.
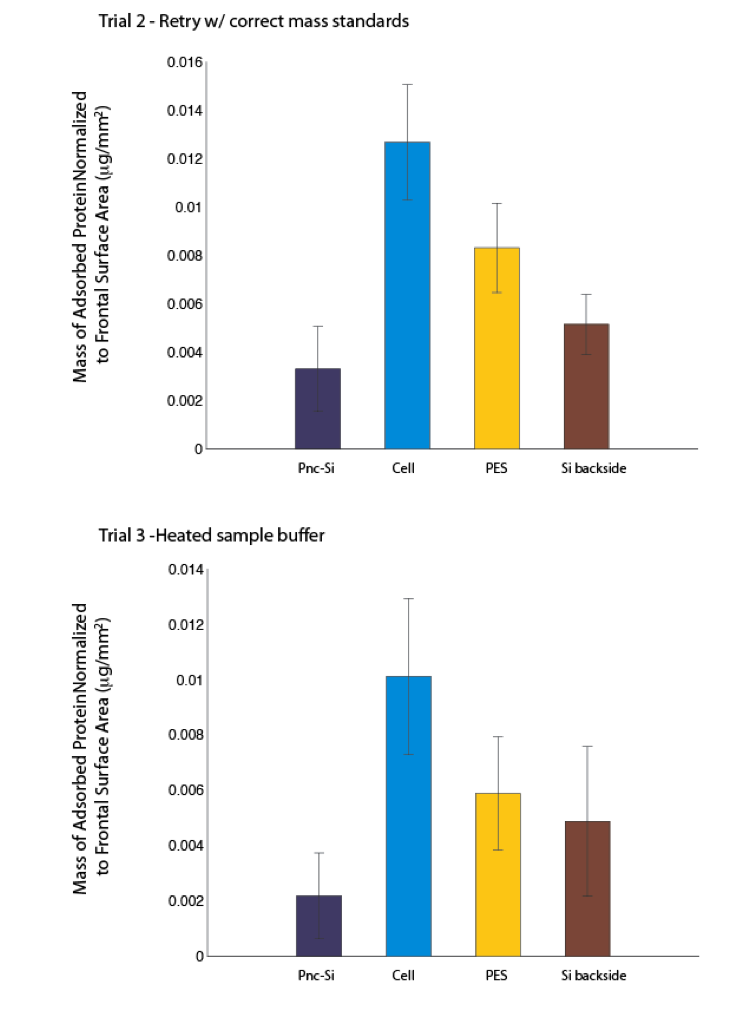
I went back and remade the standards and carefully reran 4 gels (pnc-Si, single side cellulose, single side PES, crystalline silicon). This is the upper figure. I then again reran these 4 gels using boiling sample buffer in the method. This is the bottom figure. I was surprised that the boiling sample buffer preparation actually had less overall protein in the adsorption. There isn’t any real change in the relative adsorption, so I don’t think boiling is necessary and the difference may only be in the error of the gels (feel free to argue this point).
Here’s the most important points from these retrials:
- Single side cellulose and PES adsorb more protein than pnc-Si and crystalline silicon (which should both be similar to glass).
- Cellulose adsorbs more than PES.
Both of these points are not accepted by the field. As we discussed in journal club and as indicated by Millipore, cellulose should adsorb less than PES. Also these membrane materials are made to be less sticky than glass. So is the problem with my hands or the inherent error with this assay, or are we achieving real but potentially disastrous results here?
If anything, it would seem like your assay UNDER estimates the amount of bound protein, since you may not get it all off to measure. As long as the application of each protein reflects the reality of how each device is used, it seems fine to me. That said, I suppose someone could argue that sample buffer removes protein more efficiently from different materials, so your results could be skewed. Is there any way to confirm that there is no bound protein left on the devices and it has efficiently been captured by the samples buffer?
Is there any way to keep track of all the protein that you use? Maybe apply a dilute solution, then remove it and measure the concentration to see how much has been lost. Then rinse and see if any more comes off and quantify it. It seems like a simple ELISA could provide this info. It’s more expensive, but it could confirm your results. You may need to play with the optimal initial solution concentration…